
Data Integrity in the Pharmaceutical Industry

From day one,
agile compliance
that empowers
your team!
M otivated by an increase in the number of nonconformities in the pharmaceutical industry, regulatory agencies such as FDA, MHRA, WHO and PIC/S have issued guidance in recent years on how to keep intact critical data related to the production of their products. During inspections, regulatory bodies have observed an increase in GxP breaches involving data integrity.
Problems related to data integrity have led to risks to the safety, efficacy, and quality of the products, associated with the breach of trust of regulatory agencies, patients and company shareholders.
Data that must maintain ‘preserved integrity’
All final data regarding the production process, metadata, which carry information about the main data and raw data should be a part of the integrity assurance process.
The first action that must be considered in the search for data integrity is to record the current situation of the company with an analysis of compliance with the regulatory standards in the management of critical data.
Subsequently when preparing risk analysis, it is necessary to define which data is related to good practices and which is related to the release of the batches of medicines.
The specification of the data that must be entered in this process is intended to prevent all company data, even those that aren’t subject to regulatory standards, from being included in the integrity assurance process.
If this is not done, it would increase costs and bureaucracy of the production process and delay the release of the data in the Quality Control analysis results.
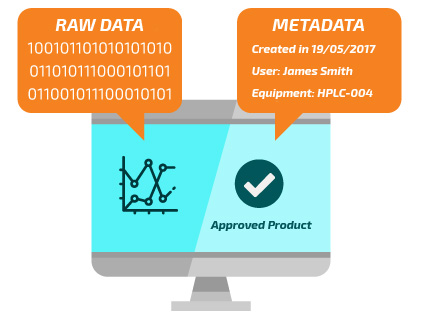
Main Data
RAW Data
Metadados
According to the GAMP5® Guide, raw data is all data on which quality decisions are based.
Some examples of raw data are photographs which accompany product complaints; environmental monitoring; calibration records; equipment qualification; equipment cleaning; maintenance records; batch production records; manual transcripts of displays and equipment; HPLC chromatograms; inventory records; and training records.
Master plans and procedures should consider this information so that the Quality System has clearly defined which data will be subject to the data integrity best practices.
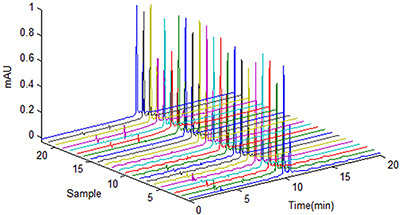
Exemplo de dados brutos obtidos de uma cromatografia.

Data Integrity Characteristics - FDA
The ALCOA acronym is a concept of data integrity based on the accurate, complete, and consistent recording and management of a data or information, either on paper or electronically. The term refers to the characteristics of data integrity which means that the data according to the FDA must be:
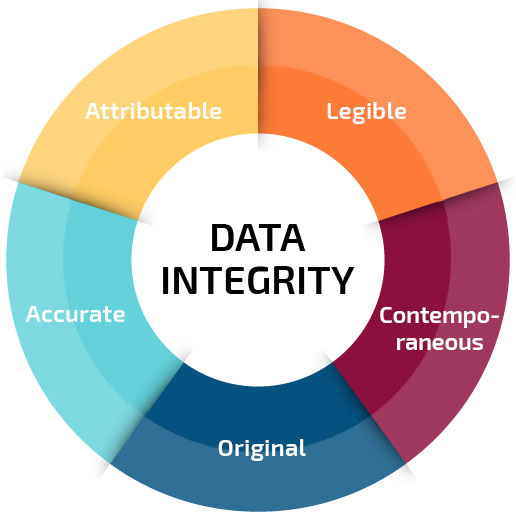
Attributable: the generated or collected data must be attributed to whoever performed the action and when the action was performed. This assignment can be done manually, through a signature or electronically dated by Audit Trail.
Legible: the collected or generated data must be registered in a legible and permanent way.
Contemporaneous: the data must be registered at the moment that the action happened.
Original: the data must be directly noted in the official log book of the company, avoiding any changes and the subsequent transcription of the data.
Accurate: the generated data must be free of errors, complete and authentic, reflecting exactly what the determined actions in the production process were.
ALCOA + PIC/S Concept
Complete
Consistent
Enduring
Available
How the process can contribute to critical data integrity
Some actions may be taken in the production process and the execution of procedures that contribute to the requirements of data integrity of regulatory agencies. These measures ensure that processes are defined so that critical data is generated and protected against changes.

Keeping vigilant in the activity area avoids inaccurate time annotations. An action that contributes to the recording of the data should be accurate.

Controlling access to computerized systems prioritizes the traceability of the data and ensures that it is attributable.

Facilitating operator access to activities-related documents that are carried out, avoids the registration of data after the action, determining that the data isn’t current.

Avoiding transcription of data, using printers directly connected to equipment ensures that the data is original.

Controlling unfilled forms is one way to minimize the risk of entry errors and prevent data from being inaccurate.

Facilitating access to raw data by those responsible for reviewing the data, minimizes the risk of indication of nonconformities by regulatory agencies.
How regulatory bodies and industries address the issue
Regulatory Agencies conduct audits focused on data integrity and they are committed to tracking fraud cases around the world.
Regulatory bodies share cases of nonconformity among themselves to get a global picture of how the issue has been treated by the industry.
For industries, it is essential to invest in employee training to deal with relevant GxP data and to create an on-site culture of attention to procedures that involve the integrity of critical data so that employees feel responsible for the integrity of the data generated in their area, production stage or analysis.
Technology has emerged as the greatest ally of transparency and a fundamental tool for increasing data reliability and integrity.

It’s now possible to manage and execute validation/qualification projects using the Digital Compliance Platform (DCP) GO!FIVE® where the specialist's efforts will be assessing vulnerable risk scenarios including mitigations as well as data integrity items where applicable.
The DCP contains pre-ready validations and data integrity assessments, which include metadata and raw data that are considered GxP relevant.
In computerized systems validation projects, it’s possible to easily import risk scenarios, requirements, and test scripts from the library that will bring items related to data integrity, access control, audit trail, and electronic signature, among others to evaluate the process and data related to systems and/or processes.
GO!FIVE® contributes to increasing compliance as it complies with FDA 21 CFR Part 11, as well as enabling data integrity projects within the system, including the evaluation of paper and electronic records from the client's site.
- Modernize and facilitate inspection and access to data related to validation/qualification projects
- Automatic and real-time tracking of all changes
- History of changes by comparison
- A knowledge base that facilitates the sharing of global best practices
- Team empowerment
- End-to-end integrity assurance on projects
- Alert of previous strange behaviors, such as simultaneous access attempts, and execution of documents in a different order from the reference guides
- Analyze data in real-time and determine the best way to track and mitigate their risk scenarios
- The Digital Compliance Platform does not allow retroactive signature
- DCP does not allow the exchange of evidence after test execution approval
- Alert or block test script execution with related requirement and/or risk scenario with pending review/approval
- Compliance with:
- FDA 21 CFR Part 211, 68, 188, and 192
- EudraLex Vol. 4, Chapter 4, Guidelines
- EudraLex Vol. 4, Annex 17, Real-time release testing and parametric release
- MHRA Guidance on GxP data integrity
- WHO Guidance on good data and record management practice
If you would like to learn more about the software that contributes to increasing data integrity in your company, please contact our experts at [email protected]
REFERENCES:
EMA: Questions and answers: Good manufacturing practice - Data Integrity Section – Ago/2016;
FDA: FDA Data Integrity Guidance – Dez/2018;
MHRA: MHRA Data Integrity Definitions and Guidance – Mar/2018;
PIC/S: PIC/S Guidance – Jul/2021;
WHO: WHO Guidance on data integrity, WHO TRS 1033, starting on page 135, 2021
GAMP5® is a guide that has its intellectual rights reserved by ISPE™. Available for purchase at ispe.org
Article written with the collaboration of Demetrius Rocha. Translated by Alissa Freire and review by Silvia Martins, Technical Director of Five Validation.



{kind=link}
{kind=link}
{kind=link}